Sarcoidosis
Salient features
·
A systemic granulomatous disorder of unknown origin that most
commonly involves the lungs
·
Cutaneous manifestations of sarcoidosis are seen in up to
one-third of patients and may be the first clinical sign of the disease
·
Red–brown to violaceous papules and plaques appear most often on
the face, in particular the nose, neck, upper back and extremities, as well as
within scars and tattoos
·
Erythema nodosum is the most common nonspecific
inflammatory skin finding that may be associated with an acute form of
sarcoidosis which tends to remit; it suggests a good prognosis.
·
Histologically, sarcoidosis is characterized by non-caseating epithelioid
granulomas, usually with a sparse or absent surrounding lymphocytic
inflammation (i.e. “naked” granulomas)
Introduction
Sarcoidosis
is an antigen‐mediated multisystem granulomatous disease of unknown etiology
characterized by the presence of non‐caseating
epithelioid cell granulomas in multiple organs. It involves mainly the lungs,
mediastinal and peripheral lymph nodes, eyes and skin. Less frequent but
usually severe manifestations can occur in the liver, spleen, central nervous
system, heart, upper respiratory tract and bones. Cutaneous lesions of sarcoidosis
may be specific, showing histopathologically sarcoid granulomas, or non‐specific, do not show sarcoid granulomas. Cutaneous
involvement in sarcoidosis is important for several reasons. Skin involvement
may be the presenting sign of systemic sarcoidosis. Skin biopsy is easy to perform,
enabling early diagnosis. Some types of cutaneous sarcoidosis have prognostic
significance and may help to predict the outcome of the systemic disease.
Epidemiology
Age
Sarcoidosis, which occurs in
patients of all races and ages as well as both sexes, is characterized by a
bimodal age distribution, with peaks between 25 and 35 years and then between
45 and 65 years in women.
Sex
Sarcoidosis is more common in women than men. In the
subpopulation with Löfgren syndrome there is a clear predominance in women.
Associated
diseases
An increased risk of developing
lymphoproliferative diseases, mainly Hodgkin lymphoma, has been reported in
sarcoidosis. There is concomitant association of sarcoidosis with a number of
autoimmune diseases, including Sjögren syndrome, systemic sclerosis, rheumatoid
arthritis, vasculitis, psoriasis, autoimmune chronic hepatitis and primary
biliary cirrhosis. Autoimmune thyroid disease, particularly Graves’s disease
and Hashimoto thyroiditis, has been associated with sarcoidosis as well.
Genetics
A positive family history of
sarcoidosis ranges from 2.7 to 17% and having a first‐degree relative with sarcoidosis increases the risk of
disease fivefold.
Etiology
and pathogenesis
Exact
etiology is unknown, but it is not an infectious disease.
Sarcoidosis
represents an exaggerated immune response to pathogen-associated molecular
patterns of dead or partially degraded mycobacteria and propionibacteria, but
also to other organic and inorganic substances.
Contemporary
concept:
•
Exposure of individuals with genetically determined “susceptible” to specific
environmental agents
Hence,
a co-existence of at least 2 factors is needed for sarcoidosis to develop:
•
An individual with “sarcoid constitution” (e.g. association with HLA-DQB1,
Iannuzzi et al. 2003)
•
Exposures to infectious agents (viruses: herpes virus, Epstein-Barr,
retroviruses; bacteria: Propionibacterium acnes, Borrelia burgdorferi,
Mycoplasma, Chlamydia, non-tuberculous mycobacteria and cell wall-deficient
mycobacteria; antigens of Mycobacterium tuberculosis: katG, Heat shock protein
(Hsp) 70, mycolyltransferase antigen 85A)
However, the absence of caseation
necrosis, the negativity of purified protein derivative (PPD), and the lack of
response to antituberculous treatment are arguments against mycobacterial
involvement in sarcoidosis.
Immunopathogenesis
Sarcoidosis
is a multisystem granulomatous disease characterized by hyperactivity of the
cell-mediated immune system. Patients with a genetic susceptibility are exposed
to a triggering antigen, leading to activation of macrophages and T cells, with
subsequent granuloma formation. While classically considered a Th1-predominant
immune response, the inflammatory cascade in sarcoidosis likely spans multiple
pathways, including the innate immune system (via activation of pattern
recognition receptors such as Toll-like receptors or NOD-like receptors) and
potentially the Th17 arm of the immune system. Specifically, upregulation of
CD4+ T helper cells of the Th1 subtype occurs
following antigen presentation by monocytes bearing MHC class II molecules,
which initiates formation of epithelioid granulomas in a variety of tissue
types. In the lung, an oligoclonal α/β T-cell population has been described, suggesting
that antigenic triggers of sarcoidosis favor a progressive accumulation and
activation of specific T-cell clones.
Increased
production of Th1 cytokines, including interleukin (IL)-2, IL-12, IL-18 and
interferon (IFN)-γ, as well as release of tumor
necrosis factor (TNF)-α by macrophages and
some CD8+ T cells, leads to persistent Th1 activity and
persistent IFN-γ elevation. There is
macrophage accumulation and hyperactivity, along with B-cell stimulation and
hypergammaglobulinemia. TNF-α and GM-CSF
promote fusion of activated macrophages into the multinucleated cells seen
within the granuloma.
IFN‐γ
activates macrophages and induces transformation into giant cells while TNF‐α induces its differentiation into epithelioid cells. Macrophages
also release chemokines such as CXCL10, attracting additional T cells of
CD4/TH1 phenotype. It is postulated that IFN‐γ
inhibits apoptosis in macrophages through the expression of high levels of P21,
which leads to granuloma perpetuation. TNF‐α
is considered the main cytokine in the development and maintenance of the
granuloma, and it is considered the cause of pulmonary fibrosis by stimulating
fibroblast proliferation and collagen synthesis. For these reasons, it is
considered that anti‐TNF‐α agents might be beneficial in sarcoidosis.
Monocyte
chemotactic factor (MCF), produced by activated T helper cells, attracts
monocytes from the circulation into peripheral tissues. Compartmentalization of
granuloma-forming T lymphocytes and monocytes within peripheral tissues leads
to lymphopenia and decreased delayed-type hypersensitivity to common antigens
(anergy), most pronounced during the initial stages of sarcoidosis. In
addition, T regulatory cells may play a role in this anergic state. However, in
some patients, there actually may be inadequate regulatory T-cell function such
that production of TNF-α or IFN-γ is not suppressed.
A clinically important aspect of
the pathology of sarcoidosis involves the development of fibrosis. Dense band
of fibroblasts may encase the ball-like granulomas. The fibrotic response can
produce tissue destruction and organ dysfunction that is often irreversible.
Currently available chemotherapeutic agents for sarcoidosis can effectively
treat the granulomatous inflammatory response but not the fibrotic reaction.




Pathology
The histopathological changes are
similar in all organs affected by sarcoidosis. The cardinal feature is the
sarcoid granuloma, defined as aggregates of epithelioid cells surrounded with a
sparse lymphocytic component, the so‐called
‘naked granuloma’. Central caseation is usually absent, although fibrinoid deposition
may be observed in up to 10% of cases. In
the skin, sarcoid granulomas are usually observed in the dermis but can also
extend to subcutaneous tissue. Multinucleated “giant cells” that results from fusion of
epithelioid cells, are usually of the Langhans type,
with nuclei arranged in a peripheral arc or circular fashion. The giant cells
may contain eosinophilic stellate inclusions known as asteroid bodies or
rounded laminated basophilic inclusions known as Schaumann bodies, although
neither is specific or required for the diagnosis. Asteroid bodies represent
engulfed collagen, whereas Schaumann bodies likely represent degenerating
lysosomes. Notably, up to 20% of biopsies of sarcoidosis contain polarizable
material; therefore its presence does not exclude the diagnosis. In vulvar
sarcoidosis, transepidermal elimination of the granulomas may be seen.
In subcutaneous sarcoidosis, the
granulomatous infiltrate is limited to subcutaneous tissue and is mainly
lobular with appearance of granulomatous lobular panniculitis, sometimes with
intense fibrosis.
Clinical
features

Systemic manifestations of sarcoidosis
Not infrequently, sarcoidosis is
discovered by chance on a chest radiograph. The clinical onset of sarcoidosis
may be acute or insidious. Acute or subacute sarcoidosis develops over a period
of weeks or a few months and it usually heralds a good prognosis. It is
characterized by mild constitutional symptoms such as fatigue, malaise,
anorexia, weight loss, low‐grade fever, arthralgia and
respiratory symptoms. An insidious onset for several months is usually associated
with respiratory complaints without constitutional symptoms, or with symptoms
referable to organs other than the lung. It correlates with a chronic course
and permanent organ damage.
Pulmonary sarcoidosis
Lung disease occurs in ~90% of patients, ranging from alveolitis
to granulomatous infiltration of the alveoli, blood vessels, bronchioles,
pleura, and fibrous septa. The end stage of pulmonary sarcoidosis is fibrosis
with bronchiolectasis and “honeycombing” of the lung parenchyma. Hilar and/or
paratracheal lymphadenopathy, which is usually asymptomatic, occurs in 90% of
patients. Patients with pulmonary sarcoidosis
are often asymptomatic, with the disease detected on a screening chest
radiograph. Common symptoms include dry cough and dyspnea on exercise. Hemoptysis
is rare. Pulmonary sarcoidosis is classically divided into four stages on the
basis of the chest radiograph.
Chest
radiograph stages in pulmonary sarcoidosis
|
Chest
radiograph stages |
%
at onset |
%
with resolution |
|
Stage 0:
normal chest radiograph |
<10 |
– |
|
Stage I:
bilateral hilar lymphadenopathy without pulmonary involvement |
50 |
60–90(<10%
progress to pulmonary involvement) |
|
Stage II:
bilateral hilar lymphadenopathy with pulmonary involvement |
30 |
40–70 |
|
Stage
III b: pulmonary involvement without bilateral hilar
lymphadenopathy |
10–15 |
<10–20 |
|
|
|
|
b |
Stage III may
be sub classified into stage IV, which includes cases with advanced pulmonary
fibrosis (hilar retraction, coarse linear opacities, honeycombing, bullae,
emphysematous changes, architectural distortion and pulmonary hypertension). |
In sarcoidosis patient with normal
lung parenchyma on chest radiograph, pulmonary function tests are abnormal in
20-40% cases. When the chest radiograph is abnormal, pulmonary function tests
are abnormal in 50-70% cases
Extrapulmonary sarcoidosis
Eye
Ocular involvement occurs in 15–20% of patients. Eye
involvement with sarcoidosis is potentially vision threatening and the patient
may be asymptomatic. For this reason, every patient diagnosed with sarcoidosis,
slit‐lamp and ophthalmoscopic examinations should be performed. Any
portion of the eye may be involved, and eye involvement may be the first
manifestation of the disease. Uveitis is the most common ocular manifestation
and can lead to cataracts and glaucoma. Other ocular manifestations include
conjunctivitis, lacrimal gland involvement causing keratoconjunctivitis sicca
(dry eyes), and optic neuritis, which may rapidly lead to a loss of vision. Heerfordt
syndrome includes fever, parotid gland enlargement, facial palsy, and anterior
uveitis.
Reticuloendothelial
system
Peripheral
lymphadenopathy involving the cervical, supraclavicular, epitrochlear, axillary
and inguinal nodes may be present. In addition to intrathoracic nodes,
mesenteric chain and retroperitoneal lymph nodes may be involved. Splenic
involvement is frequent, although splenomegaly occurs in only 5–10% of cases,
and may result in hypersplenism and pancytopenia. Bone marrow involvement is
rare.
Liver
Mild
hepatomegaly with slight cholestasis occurs in 20–30% of patients. Non‐caseating granulomas are present in up to 75% of liver
biopsies. Hepatic sarcoidosis affects the periportal areas. An increased serum
alkaline phosphatase level occurs in approximately one third of patients.
Rarely, hepatic sarcoidosis may lead to a primary billiary cirrhosis-type
picture, and portal hypertension may develop.
Neurosarcoidosis
Any
portion of the CNS or PNS may be affected by sarcoidosis. Five to 10% of
patients with sarcoidosis have clinically recognizable neurological
involvement. The disease has a predilection for the basal meninges, so cranial
nerve involvement, particularly facial paralysis, is common. In fact, it is
common for Bell palsy to be first manifestation of sarcoidosis and resolve
completely before other manifestation of the disease occur. Mass lesions may
develop in the brain or spinal cord. Aseptic meningitis and peripheral
neuropathy are other neurological manifestations of sarcoidosis.
Musculoskeletal
system
Transient
or chronic polyarthralgias are common but frank arthritis is uncommon.
Asymptomatic muscle involvement is also common but symptomatic diffuse or
nodular myopathy is rare. Bone lesions, usually osteolytic, are not frequent
and when present are located predominantly on the hands and feet.
Heart
Clinical
cardiac involvement occurs in 5% of patients, although myocardial granulomas
have been found in approximately 25% of patients at autopsy. Most clinical
problems are related to cardiac arrhythmias or left ventricular dysfunction.
Sudden death may occur. Congestive heart failure may result when the myocardium
is massively infiltrated with granulomas. Because of the potential lethal
nature of heart involvement with sarcoidosis, an electromyogram is recommended
for every patients diagnosed with the disease.
Other
manifestations
Parotid
involvement is frequent and may produce parotid enlargement, usually bilateral,
and xerostomia. The sinuses and the
upper airway are commonly involved with sarcoidosis, a condition known as SURT:
sarcoidosis of the upper respiratory tract. Nasal SURT is often associated with
lupus pernio skin lesions and may cause epistaxis and severe nasal crusting.
Sarcoidosis
may also cause a disorder in calcium metabolism that result from activated
sarcoidal macrophages demonstrated an increase in 1-alfahydroxylase activity.
This enzyme converts 25 hydroxy vitamin D to 1, 25dihydroxy vitamin D, the
active form of the vitamin, an increase of which may result in hypercalcemia,
hypercalciuria and nephrolithiasis. Sarcoidosis may be associated with a
reduction in any blood cell line. Leucopenia may result from bone marrow
involvement or from splenic sequestration. Thrombocytopenia may result from
bone marrow involvement, splenic sequestration or from an idiopathic
thrombocytopenic purpura-like syndrome related to hypergammaglobulinemia often
seen in patients with sarcoidosis.
|
SYSTEMIC
MANIFESTATIONS OF SARCOIDOSIS |
|||
|
Organ |
% of patients affected |
Clinical
manifestations/radiographic and laboratory findings |
Evaluation |
|
Lungs |
90–95 |
Dyspnea,
non-productive cough/pulmonary infiltrates, fibrosis, restrictive lung
disease (↓VC, ↓RV, ↓TLC, ↓DLCO) |
CXR, high resolution
chest CT scan (more sensitive than CXR), PFTs that include DLCO |
|
Lymph nodes (LN) |
30–40 |
Lymphadenopathy/enlarged
hilar and/or paratracheal LN |
CXR, high resolution
chest CT scan (more sensitive than CXR) |
|
Eyes |
25 |
Uveitis (can be
asymptomatic despite being severe), conjunctivitis, sicca symptoms |
Yearly ophthalmologic
examination |
|
Liver/spleen |
10–20 |
Hepatomegaly and/or splenomegaly
(rarely clinically relevant), cirrhosis, consequences of splenic enlargement
(e.g. thrombocytopenia)/↑LFTs, ↓platelets |
·
LFTs,
physical examination ·
If
clinically relevant hypersplenism suspected, abdominal/pelvic CT scan
(lymphadenopathy common finding) |
|
Heart |
25 (5% clinically
relevant) |
Palpitations, sudden
death, CHF/arrhythmias, cardiomegaly |
· EKG, echocardiogram, Holter monitor ·
If
any abnormalities on history, physical examination, or initial screening,
referral to cardiologist and additional testing (PET scan, cardiac MRI) |
|
CNS and peripheral
nervous system |
10–20 |
Neuropathies –
cranial, spinal cord, peripheral, small fiber |
·
Dictated
by symptoms (e.g. MRI, nerve conduction studies) ·
Referral
to neurologist |
|
Upper respiratory
tract, including sinuses |
5–10 |
Sinusitis, nasal
congestion, stridor, parotitis |
Referral to
otolaryngologist and/or dedicated imaging |
|
Bones |
5–10 |
Usually
asymptomatic/lytic bone lesions |
Radiography |
|
Joints/muscles |
5–10 |
Arthritis, weakness
(up to a third of patients have severe fatigue), myopathy |
·
Referral
to rheumatologist ·
EMG |
|
Bone marrow |
50 |
Lymphopenia
(↓CD4+:CD8+ ratio),
leukopenia, eosinophilia, hypergammaglobulinemia, non hemolytic anemia (5%) Elevated
risk of developing lymphoma is debatable |
CBC, SPEP |
|
Kidneys |
10–40 |
Nephrolithiasis/hypercalciuria,
↓renal function |
·
BUN,
crt, serum calcium, spot urine calcium : crt ratio, 24-hour urine for calcium
excretion ·
Referral
to nephrologist |
|
Endocrine |
5–10 |
Pituitary
or thyroid dysfunction Hypercalcemia
(increased calcitriol synthesis by sarcoidal histiocytes) |
·
Thyroid
function tests ·
Expanded
hormonal testing when clinically indicated |
|
Other |
<1 (rare) |
“Masses”
– GI tract (luminal), ovarian, testicular – often asymptomatic Granulomatous
breast infiltration that can lead to ulceration |
Site-specific imaging |
CHF, congestive heart failure; CT, computed
tomography; CXR, chest X-ray; DLCO, diffusion lung capacity for carbon
monoxide; EMG, electromyography; LFTs, liver function tests; MRI, magnetic
resonance imaging; PET, positive emission tomography; PFTs, pulmonary function
tests; plts, platelets; RV, residual volume; SPEP, serum protein
electrophoresis; TLC, total lung capacity; VC, vital capacity.
Cutaneous
manifestations of sarcoidosis
Up to
a third of patients with systemic sarcoidosis develop skin lesions, which may
be the first or only clinical manifestation of the disease. Cutaneous lesions of sarcoidosis are classified as specific
and non‐specific. Specific lesions are those that
histopathologically display sarcoid granulomas. The most frequent specific
lesions are maculopapules, plaques, lupus pernio, scar‐sarcoidosis and subcutaneous sarcoidosis. The most important
non‐specific lesion is EN and does not exhibit sarcoidal
granuloma. Cutaneous lesions of sarcoidosis are more frequent in women than in
men (2: 1).
|
SPECTRUM OF
CUTANEOUS MANIFESTATIONS OF SARCOIDOSIS |
|
|
Common |
|
|
Papules – favor periorificial sites on face |
Plaques – favor trunk and extremities |
|
Lupus
pernio – favors nose
and central face |
Scar-associated
– initial insult weeks to decades prior |
|
Tattoo-associated – favors sites of red and yellow
pigments |
|
|
Uncommon |
|
|
Annular |
Atrophic
– favors head and neck region |
|
Lichenoid |
Psoriasiform |
|
Subcutaneous
(Darier-Roussy) – favors extremities |
|
|
Rare |
|
|
Alopecia
– scarring or nonscarring |
Angiolupoid – favors face and can mimic rosacea |
|
Erythrodermic |
Hypo
pigmented |
|
Ichthyosiform |
Micropapular |
|
Nail
dystrophy – subungual hyperkeratosis, onycholysis |
Photo
distributed/photo-exacerbated |
|
Ulcerative |
Verrucous |
|
Nonspecific and common |
|
|
Erythema
nodosum |
|
SPECIFIC FORMS OF CUTANEOUS SARCOIDOSIS
Specific cutaneous lesions develop
in 9–37% of patients with systemic sarcoidosis. Although they can appear at any
time, they are usually present at the onset of sarcoidosis and the diagnosis is
frequently made by dermatologists. In the initial evaluation of patients with
suspected sarcoidosis the entire skin surface must be examined. Because
cutaneous biopsy is innocuous, it can provide a rapid diagnosis of sarcoidosis
and can avoid aggressive diagnostic techniques.
The presence of specific cutaneous
lesions have some prognostic significance in the progression of sarcoidosis as
some types of cutaneous lesions are associated with acute forms of sarcoidosis with
a favorable prognosis and others with
chronic forms with a less favorable prognosis.
The clinical appearance is due to
the presence of epithelioid cell granulomas in the dermis. Specific lesions are
red‐brown or red‐violaceous in color, generally
multiple, and do not cause symptoms. Diascopy reveals the subtle yellow-brown
or ‘apple jelly’ color characteristic of granulomatous diseases but usually
more opaque than in lupus vulgaris. In addition to the color change, the
underlying granulomas have a nodular quality that can also be appreciated with
diascopy. The epidermis rarely appears clinically involved, but often the
lesions have a waxy appearance, which reflect mild epidermal atrophy. Specific lesions
favor the face, neck, upper trunk and extremities, but may occur symmetrically
or asymmetrically on any part of the skin and mucosa. Almost all morphologies have been reported, including
macules, patches, papules, plaques, and nodules. Diverse types of lesion may
coexist in the same patient.
Sarcoidosis,
maculopapular
Macules and papules are the most
common specific lesions. The firm 2-5mm papules often have a translucent red‐brown or yellow‐brown
appearance. They are usually located on the face, mainly around the eyes and in
the nasolabial folds, although the occipital area of the neck,
trunk, extremities and even mucous membranes may be involved. They are usually
transient and appear to herald the onset of the disease. Plaques and nodules
may develop from these papular lesions and may or may not retain the classic
translucent quality of the papules.
Maculopapular lesions often resolve
either spontaneously or with treatment in less than 2 years without significant
scarring. They are commonly associated with acute forms of systemic sarcoidosis
such as hilar lymphadenopathy, EN, acute uveitis, peripheral lymph nodes and
parotid enlargement. Consequently, maculopapular sarcoidosis is associated with
a more favorable prognosis than other forms of cutaneous sarcoidosis.
A particular type of papular lesion
involving the extensor surface of the knees has been reported. The papules are
grouped over the knees, frequently with a linear arrangement that confers a
lichenoid appearance. Polarizable foreign bodies are present in a high
proportion of biopsies. These lesions are usually transient and may easily be
overlooked. For this reason, the knees should always be examined when
sarcoidosis is suspected.
Sarcoidosis,
nodular and plaque
This is almost as common as
maculopapular sarcoidosis. It usually presents as multiple, round or oval,
infiltrated reddish‐brown plaques. They are larger than 10 mm in diameter,
tend to be thicker and more indurated and persistent than papules and are
sometimes mammillated. Plaques can be associated with nodular dermal lesions.
They can be located on the face, scalp, back, buttocks and extremities. Plaques
can adopt an annular appearance by means of peripheral extension and central
clearing, especially on the forehead and neck.
After treatment plaques tend to
recur; when they do resolve they can leave permanent scarring. They are
associated with chronic forms of sarcoidosis including pulmonary fibrosis,
peripheral lymphadenopathy, splenomegaly and chronic uveitis. In patients with
plaque‐type lesions, the activity of the systemic disease usually
persists for more than 2 years.
Sarcoidosis:
lupus pernio
Lupus pernio is the most distinctive
manifestation of cutaneous sarcoidosis. It tends to appear in older people than
other forms of cutaneous sarcoidosis, and is especially frequent in women. Erythemato-violaceous,
relatively symmetric indurated plaques and nodules, primarily involving
areas most affected by cold (i.e. pernio), including the nose, cheeks, earlobes, and digits. Lupus perniois painless and is
more recalcitrant to treatment, and may result in severe cosmetic disfigurement.
In more than half of cases, lupus
pernio is associated with sarcoidosis of the upper respiratory tract,
especially in patients with involvement of the nasal rims and may directly
extend into the nasal sinus, leading to epistaxis, nasal crusting and sinus
bone involvement. Lupus pernio usually follows an extremely chronic course and
is also frequently associated with pulmonary fibrosis, chronic uveitis, and cystic lesions within
the bones of the distal phalanges.
When the latter occur the nails are usually dystrophic.

Scar
sarcoidosis
Cutaneous sarcoidosis occurs
preferentially within scar tissue, at traumatized skin sites, and around
embedded foreign material such as silica. Tattoo sarcoidosis may be considered a variant
of scar sarcoidosis that may occur decades after tattooing and must be
differentiated from foreign‐body reactions to tattoo pigment. In
scar sarcoidosis, the old scars become inflamed and infiltrated with sarcoid
granulomas. Scar sarcoidosis may be the only cutaneous finding in a patient
with systemic sarcoidosis; therefore, it is important to closely examine scar
tissue in patients suspected of having the disease. However, more commonly it
is associated with long lasting pulmonary and mediastinal involvement, uveitis,
peripheral lymphadenopathy, bony cysts and parotid infiltration. It has been
hypothesized that foreign material frequently present in scars can act as an
antigenic stimulus for the induction of granulomas.
Subcutaneous
sarcoidosis
In subcutaneous sarcoidosis, sarcoid
granulomas are limited to subcutaneous tissue. It has been observed in 1.4–6%
of patients with systemic sarcoidosis and represents 12% of specific cutaneous
lesions. Most cases occur in women, mainly in the fifth and sixth decades of
life. Subcutaneous
nodular sarcoidosis is also called Darier-Roussy sarcoidosis. Multiple indurated subcutaneous nodules are located
principally in the extremities. The lesions are painless, non tender oval, flesh-colored
or violaceous nodules that are 0.5-2 cm in diameter and covered by normal‐appearing
skin. Some studies highlight that in most patients the lesions involve the
forearms and tend to be fusiform. The subcutaneous lesions may form indurated
linear bands from the elbow to the hand. In some cases, the dorsa of the hands
are infiltrated and the fingers develop asymptomatic firm fusiform swelling
(sarcoid dactylitis).
Subcutaneous sarcoidosis usually
appear in
the beginning of the disease and is
associated with stage I changes on chest radiograph and with less than 2 years'
activity of systemic sarcoidosis. In some patients, the nodules resolve
spontaneously.
Sarcoidosis,
other
Angiolupoid
sarcoidosis
This
is a variant of lupus pernio with prominent large telangiectatic venules. It
typically presents in women as a single raised plaque on the bridge of the
nose, central face, ears or scalp. It has been observed in 8% of patients with
cutaneous sarcoidosis in an Indian series.
Hypo
pigmented sarcoidosis
Hypo pigmented, well‐demarcated, round to oval patches are observed mainly on the
limbs. Erythematous papules can be found in the center of some lesions, leading
to a ‘fried egg’ appearance. The presence of an interface dermatitis associated
with sarcoidal granulomas may explain the hypomelanosis.
Lichenoid
sarcoidosis
This
is more frequent in children and is estimated to account for 1–2% of cases of
cutaneous sarcoidos. Multiple 1–3 mm, flat‐topped
or dome‐shaped erythematous or skin‐colored
papules may involve extensive areas of the trunk, limbs and face. Wickham
striae are absent.
Ulcerative
sarcoidosis
This
usually develops in papulonodular or atrophic lesions on the lower legs and
heals with scarring. It has been reported in 1.1–4.8% of patients with
cutaneous sarcoidosis.
Psoriasiform
sarcoidosis
Well‐demarcated erythematous scaly plaques that may be clinically
indistinguishable from psoriasis are found in 0.9% of patients with
sarcoidosis. However, psoriasis plaques have a redder color and larger scales,
and heal without scarring.
Verrucous
sarcoidosis
This
presents as well‐demarcated hyperkeratotic papillomatous lesions usually located
on the lower extremities. Most patients have longstanding systemic disease.
Necrobiosis‐lipoidica‐like lesions
Pink
to violaceous plaques with depressed centers located on the shins may resemble
necrobiosis lipoidica. The granulomatous nature of both diseases may explain
this resemblance.
Ichthyosiform
sarcoidosis
This
is characterized by adherent, polygonal, grey or brown 0.1–1 cm scales
most commonly located on the lower extremities. Biopsy reveals both sarcoid
granulomas and compact orthokeratosis with a diminished granular layer,
mimicking ichthyosis vulgaris.
Erythrodermic
sarcoidosis
Slightly
infiltrated, erythematous plaques coalesce over large areas. In contrast to
classical erythroderma, some areas of skin are spared. Some patients with
prominent scaling have been reported as acquired ichthyosiform erythroderma. As
in other atypical forms of sarcoidosis, histopathological evaluation may be
necessary to exclude other more common causes of erythroderma.
Morphoea‐like lesions
Indurated
and atrophic plaques, usually located on the thighs of women, have been
described in sarcoidosis. Some cases show a linear distribution resembling
linear morphea. In addition to epithelioid granulomas, dermal sclerosis is
observed histopathologically.
Livedo
Sarcoidosis
may rarely present with livedo. Biopsy
specimens revealed epithelioid cell granulomas around blood vessels conditioning
luminal narrowing. Sarcoidosis with livedo is characterized by a high frequency
of ophthalmological and central nervous system involvement.
SPECIAL LOCATIONS OF SPECIFIC CUTANEOUS
LESIONS
Alopecia
Alopecia occurs with involvement of
the scalp and may be scarring or nonscarring. Scale is usually absent, although
follicular plugging may be present.
Nails
Nail
changes can be seen in sarcoidosis, including clubbing, subungual
hyperkeratosis, and onycholysis. In
advanced cases, granulomatous infiltration of the nail matrix can result in
total loss of the nail. Nail sarcoidosis is associated with bony cysts in the
underlying terminal phalanx and a chronic disease course.
Oral
Oral
sarcoidosis presenting as papules and plaques that may affect the soft mucosa,
gingival tissue, tongue, hard palate, and major salivary glands. Heerfordt’s
syndrome (uveoparotid fever) includes parotid gland enlargement, uveitis,
fever, and cranial nerve palsies, usually of the facial nerve.
Genital
In the male genitalia sarcoidosis
usually presents with testicular or epididymal masses without cutaneous
lesions. Vulval sarcoidosis is rare. It presents with semi‐translucent reddish brown papules and nodules that must be
distinguished from tuberculosis, Crohn disease, syphilis, foreign‐body reactions and lymphogranuloma venereum.
NON‐SPECIFIC CUTANEOUS SARCOIDOSIS
LESIONS
EN is the
most common non‐specific lesion of sarcoidosis (prevalence of
approximately 17%). EN is frequently the initial manifestation of sarcoidosis.
It is more frequent in young women. These patients tend to have an acute form
of sarcoidosis and resolves spontaneously within 1 year.
Löfgren
syndrome is classically described as a triad of EN, polyarthritis, and hilar adenopathy.
The adenopathy may be unilateral or bilateral hilar and/or right paratracheal
lymphadenopathy. Other symptoms include anterior uveitis, fever, ankle
periarthritis, arthralgias, and pulmonary involvement.
Childhood
sarcoidosis
Childhood
sarcoidosis is rare, and it usually presents with a triad of arthritis, uveitis
and cutaneous lesions, along with constitutional symptoms. Peripheral
lymphadenopathy is frequently present, but pulmonary involvement is less common
than in adults. If sarcoidosis is being considered in a child, it is important
to exclude Blau syndrome.
Course
and prognosis
In most patients, particularly those
with an acute presentation, the disease resolves spontaneously without sequelae
within 2–5 years. Löfgren syndrome has an excellent prognosis. Ten to 30% of
patients follow a chronic progressive course, sometimes with irreversible
fibrotic changes in spite of therapy. Occasionally, recurrence of sarcoidosis
many years after spontaneous remission occurs, particularly in patients with
Löfgren syndrome. Pregnancy is not contraindicated except in severe chronic
disease. However, there may be relapses after parturition. Mortality is less
than 5%.
Investigations
Radiologic
findings include hilar and/or paratracheal lymph node enlargement with or
without pulmonary infiltrates on chest radiography. Thoracic high-resolution CT scans are more sensitive than
radiographs in detecting parenchymal and nodal disease and may be used to
delineate active inflammation from fibrosis. Pulmonary function tests reveal
restrictive lung disease, with decreased vital capacity, residual volume, total
lung capacity and diffusing capacity. Tuberculin
skin test is negative in more than 80% of patients. PET with 18F‐fluorodeoxyglucose (18F‐FDG PET) is more sensitive than 67‐gallium scan for assessing the activity and extension of
sarcoidosis. 18F‐FDG PET/CT is mainly useful in the
detection of occult granuloma sites for biopsy and in the detection of residual
activity in patients with fibrotic pulmonary sarcoidosis.
Serologically,
elevated antinuclear antibody titers occur in ~30% of patients. The serum
angiotensin-converting enzyme (ACE) level is elevated in ~60% of patients; it
has a false-positive incidence of 10%, making it a more useful test for
monitoring disease progression than for establishing the diagnosis. Most
patients exhibit lymphopenia, with a decreased CD4+:CD8+ ratio of circulating lymphocytes. Five percent
of patients have non-hemolytic anemia, and one-quarter have eosinophilia. Both
an elevated ESR and hypercalcemia may be present.
Diagnostic
criteria
Sarcoidosis
is a diagnosis of exclusion, both clinically and histologically. In order to
establish the diagnosis, a supportive clinical and
radiological picture,
must be accompanied by the histologic presence of non-caseating granulomas in
at least one organ system with negative cultures for
mycobacteria and fungus, and exclusion of other granulomatous diseases.
Box shows the basic study protocol.
The most common biopsies are transbronchial, skin, and peripheral lymph node
biopsies. When the clinical and radiological findings are not typical,
particularly with stage 0 chest radiograph, it is advisable to obtain at least
two positive biopsies. Löfgren syndrome is so recognizable that histological
confirmation may not be necessary.
Box
Recommended basic
assessment of patients with sarcoidosis
·
History (including occupational and
environmental exposure)
·
Physical examination
·
Ophthalmological examination (slit‐lamp and ophthalmoscopic examination)
·
Chest radiograph
·
Standard hematological and
biochemistry profiles (including urine and serum calcium level, hepatic enzymes
and renal function tests), and serum angiotensin‐converting
enzyme level
·
ECG
·
Pulmonary function tests (including
spirometry and DLco)
·
Tuberculin skin test
·
Biopsies (including culture for
mycobacteria and fungus)

Management
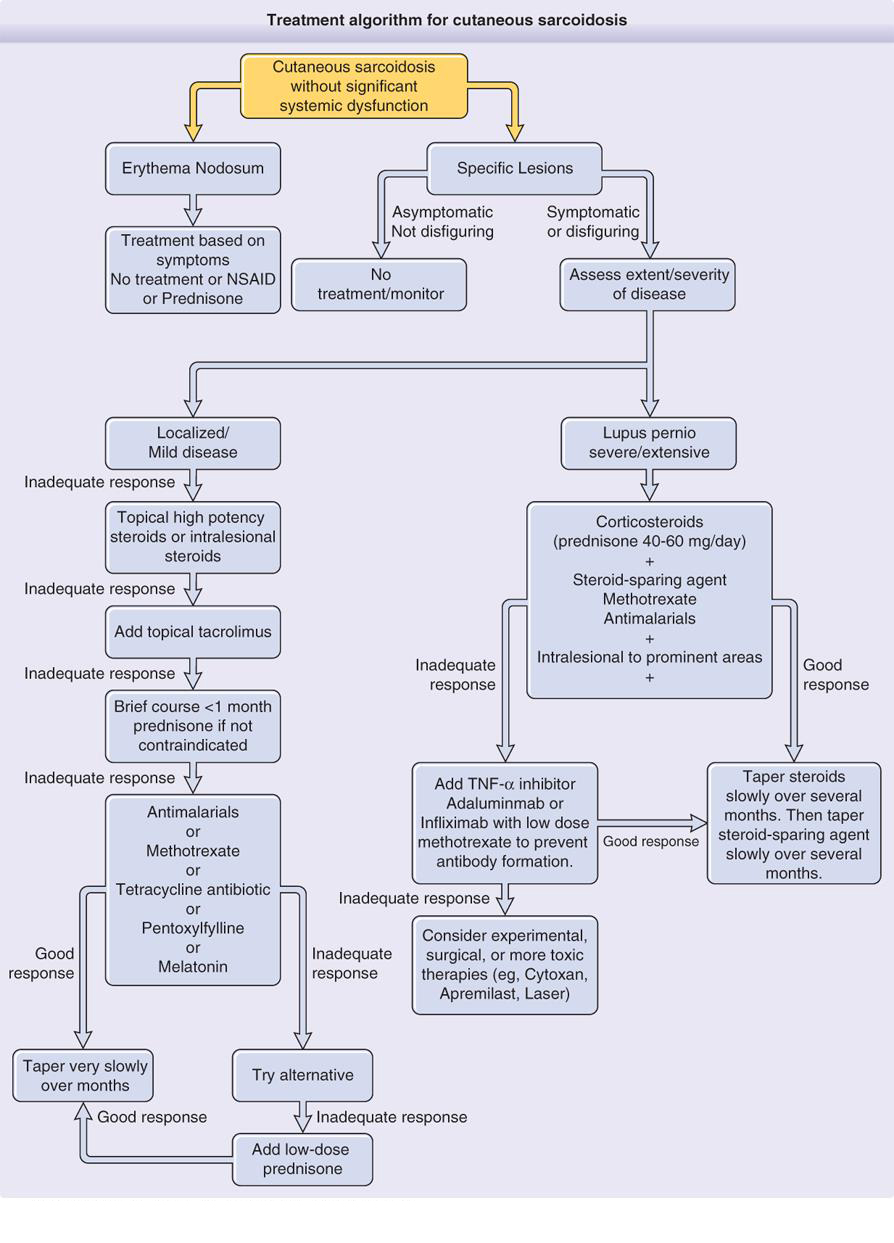
Because sarcoidosis often spontaneously remits and therapy
may be associated with significant side effects, it is not mandatory to treat
the disease. Treatment is indicated when there is evidence of progressive organ
damage.
Oral corticosteroids are the treatment of choice for active ocular
disease, active pulmonary disease, cardiac arrhythmia, CNS involvement or
hypercalcemia. The recommended dose in pulmonary
sarcoidosis is prednisolone 30–40 mg/day, with gradual reduction to
5–10 mg/day for at least 1 year. In patients with severe uveitis,
neuro sarcoidosis, or symptomatic cardiac involvement, a dose of
1 mg/kg/day can be required. The goal is alternate-day
prednisone at the lowest possible dose that will maintain a remission. When corticosteroids cannot be withdrawn, other drugs such
as chloroquine or cytotoxic drugs can be used. Most of these drugs are
inadequate as monotherapy but are effective as corticosteroid sparing agents.
For patients with cosmetically insignificant and asymptomatic
cutaneous lesions treatment may be unnecessary.
First line
Mild to moderate disease
Cutaneous lesions, including lupus pernio, may be improved
with prolonged application (longer than 8 weeks) of class 1 topical
corticosteroids, but intralesional injections of triamcinolone acetonide at
concentrations of 5–20 mg/mL repeated every 3–4 weeks are generally more
effective. Topical tacrolimus is effective in several case studies as well.
Severe disfigurement or lupus pernio
Prednisolone 20–60 mg/24 h is administered until
clinical response (usually 1–3 months) and then tapered by 5–10 mg/week to
the lowest dose that prevents relapse: corticosteroid‐sparing agents are indicated when a dose of at least
10 mg of prednisolone daily is required for this. The mechanism of
osteoporosis is multifactorial in sarcoidosis and all patients on chronic
corticosteroids should have a baseline bone density study. For patients without hypercalcemia or nephrolithiasis, oral calcium supplements may be used. The
addition of vitamin D is less clear‐cut.
Calcium levels should be checked in the summer months to detect hypercalcemia.
Bisphosphonates have been shown to be useful in treating corticosteroid‐induced osteoporosis.
Second line
Antimalarials, methotrexate or tetracycline can be used as
second line therapy for mild to moderate disease and as corticosteroid‐sparing agents in patients with severe disfigurement or
lupus pernio. They may be the first option when systemic corticosteroids are
contraindicated.
Antimalarials
Hydroxychloroquine
(200–400 mg/day) or chloroquine (250–500 mg/day) can be effective in
controlling skin manifestations of sarcoidosis, particularly chronic disease and can be used as corticosteroid‐sparing agents in severe cases. Hydroxychloroquine has a
lower risk of retinopathy but chloroquine seems to be more effective. Eye
evaluation every 6–12 months is usually recommended.
Methotrexate
Methotrexate is used either for recalcitrant skin disease or
as a corticosteroid‐sparing agent for both pulmonary and cutaneous disease.
Patients need to be monitored for neutropenia, renal function, and liver and
pulmonary toxicity. Nausea can be reduced with folic acid supplementation. Approximate
10% of sarcoidosis patients taking methotrexate develop cirrhosis, even their
LFT is normal. Therefore routine liver biopsies should be considered after 2 gm
of total therapy (usually after 2 years). Low dose methotrexate, 10-25 mg a
week, is used for the treatment of cutaneous sarcoidosis. Cutaneous improvement
may be noted within 1 month, but a maximal therapeutic benefit often does not
occur until at least 6 months after the initiation of treatment.
Tetracycline
Minocycline 100 mg twice daily is effective for chronic
cutaneous lesions. However, poor response to tetracycline has been reported in
lupus pernio. Although the mechanism of action is unclear, an anti‐inflammatory action of minocycline has been suggested.
Because of the relatively benign safety profile of tetracycline, proposed
therapeutic strategies include initiating treatment with minocycline for 3
months; if the response is unsatisfactory, hydroxychloroquine can be added, and
if the desired improvement is not achieved, methotrexate may then be added to
the regimen.
Third line
TNF‐α antagonists
TNF is a cytokine
secreted in macrophages associated with sarcoidal granuloma. Improvement of
systemic and cutaneous sarcoidosis has been observed with TNF-α inhibitors, including infliximab and adalimumab, but
these agents may also trigger sarcoidosis. They are particularly useful for the
treatment of lupus pernio.
Surgical treatment
Pulsed dye and CO2
laser treatments and photodynamic therapy may be effective for lupus pernio,
but there is also a report of laser therapy worsening the disease. Surgical
excisions with grafting have been performed for ulcerative sarcoidosis.